
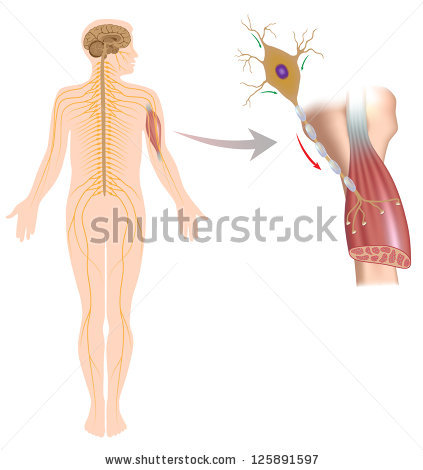
What is amyotrophic lateral sclerosis (ALS)?
Amyotrophic lateral sclerosis (ALS), sometimes called Lou Gehrig’s Disease, is a progressive brain disease that attacks the nerve cells (neurons) that control voluntary muscles. This disease belongs to a group of motor neuron disorders (e.g., muscular dystrophy, multiple sclerosis, Parkinson’s disease) that lead to the gradual degeneration and death of motor neurons, the nerve cells located in the brain, brainstem, and spinal cord. These motor neurons serve as connections from the nervous system to the muscles in the body.
Imagine wanting to brush your teeth. The motor neurons in the brain (upper motor neurons) have to send a message to the motor neurons in the spinal cord (lower motor neurons). The lower motor neurons then send the message to the muscles that are needed to brush your teeth (arms, mouth, etc.). ALS causes both the upper and lower motor neurons to degenerate or die. Both the brain and spinal cord lose the ability to initiate and send messages to the muscles in the body. The muscles, which are unable to function, gradually atrophy (waste away) and fasciculate (twitch).
ALS does not impair a person’s mind. It affects only the motor neurons. Personality, intelligence, memory, and self-awareness remain the same. The senses of sight, smell, touch, hearing, and taste remain intact as well.
What are some signs or symptoms of ALS?
ALS first affects an individual’s ability to speak loudly and clearly. Eventually, it completely prevents speaking and vocalizing. Some of the earliest speech-related symptoms of ALS may include:
- nasal speech quality (talking out of your nose)
- difficulty pronouncing words due to weak, tight, or stiff speech muscles (dysathria)
- difficulty with lengthy sentences or conversation
As muscles for breathing weaken, it becomes more difficult for patients to speak loud enough to be understood. Eventually extensive muscle atrophy eliminates one’s ability to vocalize and speak altogether.
Patients may also experience difficulty chewing and swallowing. Initially only some food consistencies are affected, but over time it becomes difficult for the patient to swallow even pureed foods and saliva. People with ALS get tired easily and they may not have the energy to eat an entire meal. All of these factors make it difficult to maintain adequate nutrition and weight. Eventually doctors may decide that it is best to insert a feeding tube into the person’s stomach to make sure they receive enough nutrition.
In some cases, initial symptoms affect only one leg or arm. Patients may have awkwardness and stumbling when walking or running. They may have difficulty lifting objects or with tasks that require manual dexterity (e.g., buttoning a shirt, tying a shoe, turning a key). Eventually the individual will not be able to stand or walk, get in and out of bed without help, or use hands and arms to perform activities of daily living, such as washing and dressing.
How is ALS diagnosed?
There is no specific laboratory test for ALS, making it hard to diagnose. The diagnosis is made using clinical findings in conjunction with results of electrodiagnostic studies and the absence of evidence of other disorders. According to the diagnostic guidelines of the World Federation of Neurology, there must be lower motor neuron degeneration detected by clinical electrophysiological or neuropathologic examination, signs of upper motor neuron degeneration by clinical examination, and progressive spread of signs within a region of the body or to other regions.
Causes of ALS
The cause of ALS is unknown, though some cases where there’s a family history of the disease are associated with mutations in the gene for an enzyme called SOD1. It’s not clear how the mutations cause motor neuron degeneration, but studies suggest the SOD1 protein can become toxic.
Scientists have identified more than a dozen other genetic mutations that may be linked to ALS. These mutations cause changes in the processing of RNA molecules (which may regulate genes), defects in the recycling of proteins, defects in motor neuron shape and structure, or susceptibility to environmental toxins.
Other research suggests ALS may share similarities to frontotemporal dementia (FTD), a degenerative disease of the brain’s frontal lobe. A defect in the C9orf72 gene is found in a substantial number of ALS patients as well as some FTD patients.
Respiratory complications
All deaths directly caused by ALS result from respiratory complications. This occurs primarily from the patient’s inability to ventilate as respiratory muscle weakness progresses. In patients with bulbar weakness, aspiration of secretions or food may occur and precipitate pneumonia, resulting in further respiratory compromise; therefore, aggressive respiratory management is necessary in the comprehensive care of patients with ALS.
Routinely measure vital capacity in the sitting and recumbent positions. Most often, the recumbent measurement declines prior to the sitting measurement. Gravity assists in lowering the diaphragm as the patient’s angle of inclination is increased. As respiratory weakness progresses, patients have increasing difficulty with diaphragmatic movement when supine because of the elimination of this effect from gravity. This results in alveolar hypoventilation and ultimate oxyhemoglobin desaturation.
Difficulty sleeping may be the first symptom of hypoventilation. Patients should be questioned routinely regarding sleep habits, and if a sleep disturbance develops, measure vital capacities sitting and supine. In addition, perform overnight oxygen saturation monitoring to assess for nocturnal hypoxemia and the need for nocturnal noninvasive intermittent positive pressure ventilation (IPPV).
Treatment for ALS
Currently, ALS does not have a cure, but treatments exist to relieve symptoms and improve patients’ quality of life.
The first drug for treating the disease, Riluzole, was approved by the Food and Drug Administration (FDA) in 1995. Riluzole is thought to decrease damage to the motor neurons by minimizing the release of the chemical signal glutamate. In clinical trials, the drug prolonged the survival of ALS patients’ (particularly ones who had difficulty swallowing) by several months. It can extend also the time before a patient must go on a ventilator.
Doctors can prescribe medications for reducing fatigue, muscle cramps, muscle spasticity, and excessive saliva or phlegm, as well as pain, depression, sleeping problems or constipation.
Speech therapists and nutritionists can help ALS patients who have trouble speaking or swallowing. As the disease advances, patients can learn to answer yes-or-no questions with their eyes or using computer-based systems.
Physical Therapy and Exercise
Physical exercise can help maintain or improve strength in the muscles not affected byALS, and maintain flexibility in muscles which are affected. It can help prevent stiffness in the joints. Physical therapy may also help people with breathing difficulties to clear their chests and maintain lung capacity. However, people living with ALS can tire very easily and find they need to conserve energy, so very strenuous exercise is not normally recommended.
Several people had found physical therapy and gentle exercise helpful, including hydrotherapy or swimming. A few had continued going to the gym and had adapted their exercise routine as their symptoms changed. Special equipment — such as ramps, braces, walkers and wheelchairs — can give patients mobility without exhausting them.
Physical therapists should instruct proper stretching and daily range-of-motion (ROM) exercises to the patient with amyotrophic lateral sclerosis (ALS) and his/her caregivers. Therapists should try to anticipate the patient’s future needs and introduce assistive devices in a timely manner. Lightweight ankle-foot orthoses can be provided to minimize foot drop and stabilize weak quadriceps muscles to prevent falls.
The physical therapist should also emphasize energy conservation and teach patients and caregivers methods for performing safe, efficient transfers. Therapists also can provide instruction for strengthening exercise programs, but exercises should be performed at submaximal levels in muscles without marked weakness and should be prescribed only for patients with slowly progressive disease.
In one study, individualized, moderate-intensity, endurance-type exercises for the trunk and limbs performed 15 minutes twice daily was shown to significantly reduce spasticity as measured by the Ashworth scale. The effects of exercise in people with ALS are not well understood. A recent Cochrane review concluded that studies were too small to determine to what extent strengthening exercises for people with ALS are beneficial or whether exercise is harmful. There is a complete lack of randomized or quasi-randomized clinical trials examining aerobic exercise in this population.
When making wheelchair recommendations, the therapist should try to anticipate the patient’s future needs. Wheelchairs should be introduced while the patient is still ambulatory in order to enhance energy conservation. Initially, a lightweight wheelchair should be rented, with plans to purchase a heavier, more expensive chair when the patient is no longer able to ambulate. Recommend modifications to the patient’s wheelchair in a timely manner and in accordance with the patient’s tolerance for gadgets.
Occupational Therapy
Occupational therapists should focus on teaching energy conservation techniques to the patient with amyotrophic lateral sclerosis (ALS) and his/her caregivers. Adaptive equipment should be introduced early and in synchronization with the patient’s progressing needs.
Upper extremity bracing can be used in cases where weakness may alter joint biomechanics. Patients with poor grasp can be provided with a universal cuff, and patients with hand weakness can be braced in 20-25° of extension to improve grip strength. For patients with proximal upper extremity weakness, a balanced forearm orthosis (ie, deltoid aid) may be beneficial to enhance upper extremity movement by eliminating the effects of gravity. When shoulder girdle weakness progresses to advanced stages, slings can be used to decrease pain by limiting traction on the associated ligaments, nerves, and vessels.
For patients with severe limb involvement, introduce environmental control units (ECUs) that utilize oral-motor movements. In patients with severe bulbar involvement, extraocular movements are usually preserved so ECUs incorporating eye gaze technology can be used.
Reference:
American Speech-Language Hearing Association http://www.asha.org/public/speech/disorders/als/
Live Science http://www.livescience.com/39583-als-lou-gehrigs-disease.html
Medscape http://emedicine.medscape.com/article/306543-overview#aw2aab6b4
Health Talk Online http://healthtalkonline.org/peoples-experiences/nerves-brain/motor-neurone-disease/physical-therapy-and-exercise